“Science is what we understand well enough to explain to a computer. Art is everything else we do.”
Donald Knuth
My research is in the world of molecular simulation. I enjoy what I do because I get to wear many hats- computer programmer, chemist, mathematician, physicist, and, very occasionally, biologist. My passion for science is motivated by curiosity and a desire to know more. Molecular simulation, at the intersection of all of these fields, is a great place to be, with interesting questions in every direction. Below are some of my research achievements so far.
Simulating a Molecular Motor
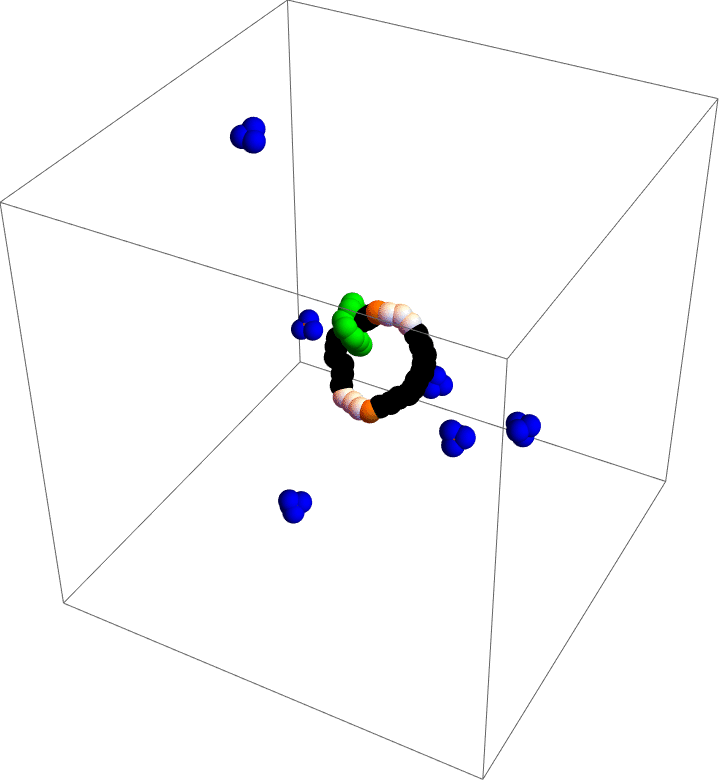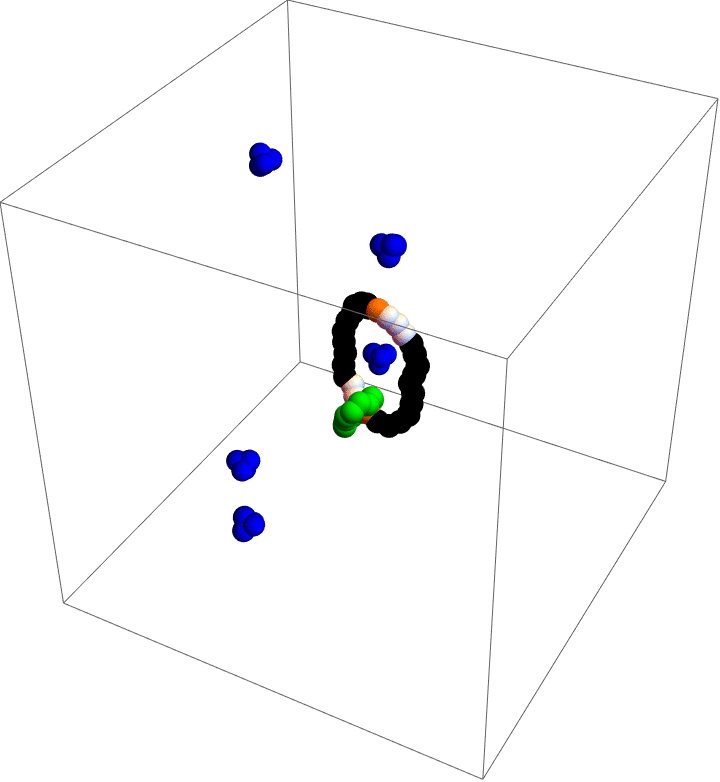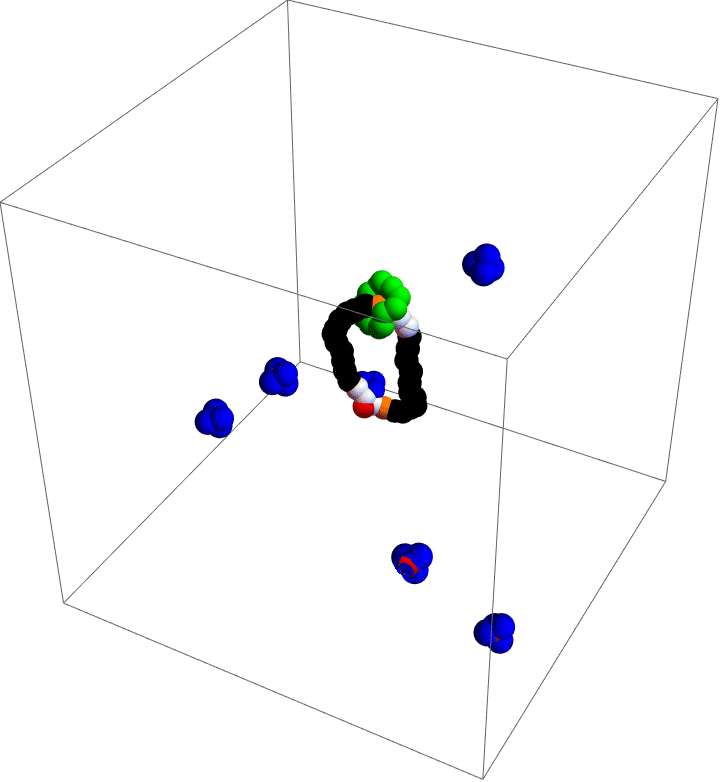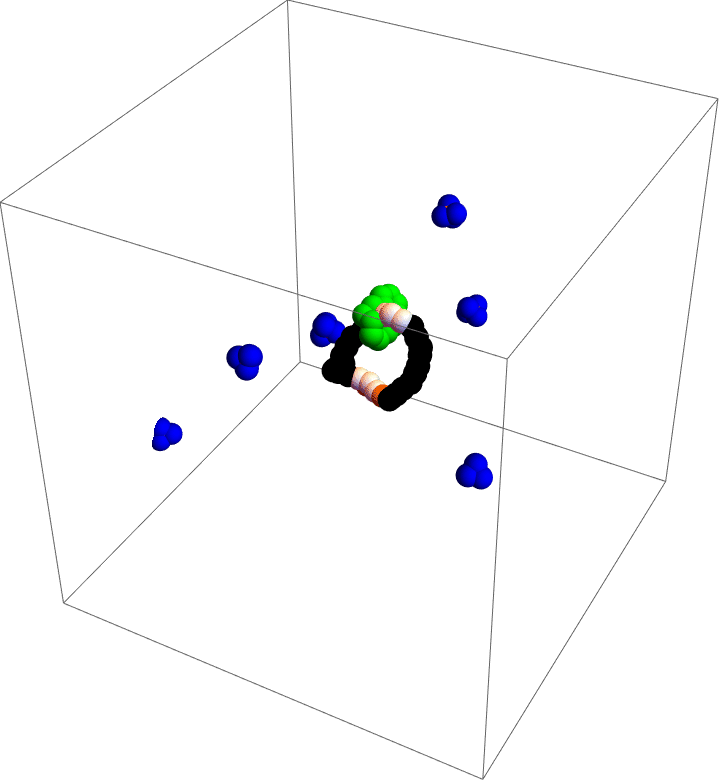
Molecular motors are incredible. As you take in food and convert that stored chemical energy into everything you do, molecular motors convert chemical energy into motion at the smallest scales in your body. Molecular simulation is a popular tool to study these diminutive machines, but studies that can capture the complete behavior of a motor often lack details and those that capture details often cannot capture the complete behavior. As a part of my postdoc work at Northwestern, I was inspired by the world’s first synthetic, autonomous, chemically-fueled molecular motor to see if computational methods could capture complete, detailed dynamics of a simple molecular motor.
My motor model (video above) consists of two interlocking rings. Fuel is made up of a red particle stuffed inside a blue tetrahedron. This structure will naturally decompose, releasing free energy. The motor catalyzes this decomposition as a way of capturing some of the free energy released and converting it into motion. This catalysis occurs at white particles in the video. Because the fuel is strongly repulsed by the small green ring, a kinetic asymmetry occurs where the fuel prefers to be catalyzed away from the green ring and not close to it. This kinetic asymmetry combined with the free energy released by the fuel create an “information ratchet” where the green shuttling ring will tend to rotate around the larger ring in a clockwise direction. As long as I maintain a nonequilibrium concentration of fuel the motor will cycle clockwise continuously, just like maintaining steady pressure on the gas pedal of a car will supply the engine with a steady stream of fuel and keep the car at a constant speed. Unlike a car, though, a molecular motor experiences relatively large random fluctuations so backward motion is always possible, too. The graph (below) shows how these clockwise (CW) and counterclockwise (CCW) rotations are unbalanced throughout a simulation of the motor. We could attach a load to this green ring now and produce useful work from the fuel’s chemical energy.

This approach has allowed me to study how fuel concentration affects the speed, accuracy, and efficiency of the motor. I can also explore how changes to the motor’s interactions or configuration affect its performance. If you’d like to read more, the details are here. This project in particular has inspired my vision for future research. I think that molecular simulation offers a way of answering lingering questions about the operation of more complex biological molecular machines.
Sampling Chemical Design Space
Determining how intermolecular interactions lead to rates is an important and ubiquitous problem in chemistry. How do different functional groups on a catalyst affect reaction rate? Or how do mutations in a protein structure affect folding rate? Computationally speaking, if I had a model of the process I could enumerate all of the possible interactions and do brute force rate calculations for each possibility, but this grid search approach is hugely expensive. As a postdoc at Northwestern, I developed a computational method that simultaneously samples intermolecular interaction space and reactive trajectory space. This means that the algorithm will wander around all of the possible interactions and all of the possible “movies” of the reaction occurring with those interactions. The algorithm is tuned to prefer areas where the reaction rate is fast. As a result, I know that the interactions I see the most often give rise to the fastest rates.

As an example, the decomposition of a red particle stuffed inside a blue tetrahedron into its components is controlled by three interaction parameters (above, top). This 3-D parameter space gives rise to different rates (above, left). My method samples parameter space in proportion to the rates (above, middle). Parameter space is sampled in proportion to the underlying rates (above, right).
You can read more about this method here. This project furthered my understanding of statistical mechanics, Monte Carlo methods, and advanced sampling techniques.
Fast & Accurate Polarization Algorithms

Electronic polarization is an important effect to capture in aqueous or heterogeneous chemical systems. But polarization is computationally expensive to calculate since the calculation usually involves an iterative procedure. In graduate school at UC Berkeley, I developed a method that dynamically integrates an auxiliary set of polarization degrees of freedom and then uses these auxiliaries to estimate the true polarization. In technical terms the method is a hybrid approach between an extended Lagrangian approach and a self-consistent field approach and applies inertial control to the auxiliaries. Naturally, then, the method was called “(inertial) extended Lagrangian/zero iteration self-consistent field”, or iEL/0-SCF for short. With this approach I can accurately capture static and dynamic properties, like the structure (above, left) and dipole autocorrelation (above, right) of liquid water without any costly self-consistent field iterations. I can get all the accuracy of including polarization, but without the cost!
If you’d like to read more, start here with an easy implementation that reduces the number of expensive iterations or here with a more complex implementation that eliminates all of the iterations. I also extended this method to the Drude polarization model here and investigated how to systematically build better estimates of the true polarization from auxiliaries here. By combining my method with a state-of-the-art integrator, I was able to do polarizable molecular dynamics with really big time steps here. I also contributed to some work that explored using parts of my method with quantum simulation here. Overall this research gave me a solid grounding with the fundamentals of molecular simulation.
